Research Interests
Spatial organization and temporal dynamics are essential properties of eukaryotic cell signaling. Eukaryotic cells are organized into membrane- and non-membrane-bound compartments, each with a specialized environment and repertoire of biomolecules tailored to its biological function. The molecular composition of these compartments is dynamically regulated in response to signals via post-translational modifications (PTMs) to proteins that direct changes in their subcellular localization. By exerting spatiotemporal control over protein function, PTMs program coordinated responses to changing conditions encountered by the cell and are a critical biological regulatory mechanism whose dysregulation often underlies human disease.
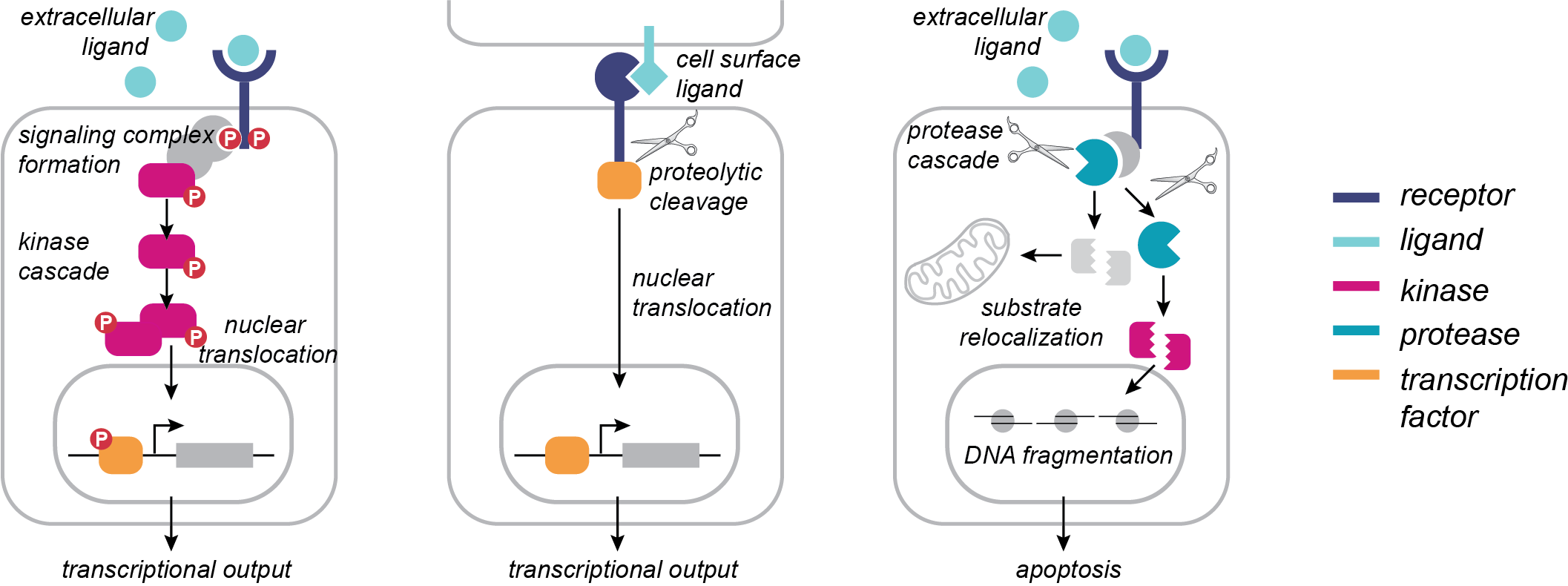
A comprehensive map of post-translationally modified proteins across subcellular space and time would transform our understanding of how PTMs orchestrate complex cellular functions in response to biological stimuli. However, current technologies are unable to provide a systems-level experimental mapping of the dynamic subcellular localization of PTM-bearing proteins. Recent improvements in mass spectrometry have enabled deep profiling of post-translational modifications (PTMs), leading to the identification of hundreds of thousands of modification sites that regulate biology. A remaining challenge is to move beyond simply cataloging these modifications to assigning their biological functions. A critical step toward this goal is to understand the spatial localization and temporal dynamics of PTMs. Our lab uses protein engineering approaches to repurpose native enzymes as chemical biology tools for covalent capture of PTMs. Two important advantages of enzymatic catalysts are 1) they can be genetically targeted to specific locations within the cell, enabling spatial resolution of PTMs; and 2) they have typically evolved to function on fast timescales in the cellular milieu, enabling temporal resolution of PTMs. Our goal is to bridge the gap between proximity labeling approaches to studying the proteome, which provide spatial information, and proteome-wide approaches for identification of PTMs, which provide clues about protein function.
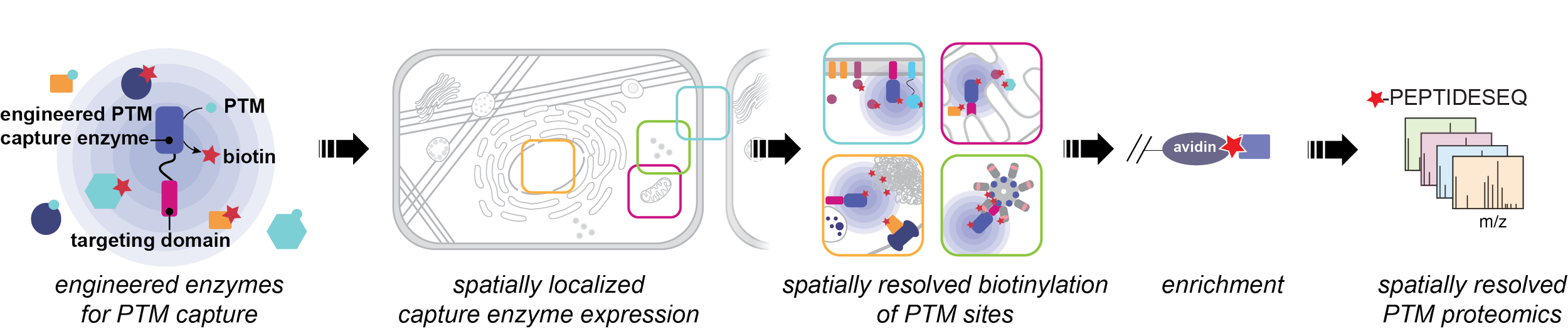
Spatially-resolved mapping of proteolysis via enzymatic N-terminal tagging
Proteolysis is a key post-translational modification that regulates a wide array of biological processes in human health and disease, including viral infection, cancer progression, organismal development, and neurodegeneration. More than 500 proteases are encoded in the human genome, comprising ~2% of the human proteome. Although the prevalence of proteases is similar to that of kinases, the identification of phosphorylation sites and elucidation of their biological roles has far outpaced our understanding of the functional roles of proteolytic cleavage events. Functionally important proteolytic cleavage events are often undetectable using conventional transcriptomics and proteomics approaches, which primarily measure changes in protein abundance. Over the past decade, technologies for the enrichment of the neo-N termini generated by proteolysis, as well as improvements in mass spectrometry, have enabled systems-level study of proteolytic cleavages. However, the challenge remains to move beyond simply cataloging these modifications to understanding their functional roles in biological regulation. We are developing and applying chemoenzymatic tools that will enable global analysis of proteolysis in defined subcellular locations.
New enzymatic tools for proximity-dependent capture of the phosphoproteome
Protein phosphorylation is a PTM that regulates a diverse array of biological processes, leading to emergent cellular behaviors such as proliferation, migration, and apoptosis. It has been estimated that more than two-thirds of human proteins are phosphorylated, and the phosphorylation signaling network is comprised of hundreds of kinases, phosphatases, and “reader” proteins that interact with tens of thousands of phosphorylation sites. Based on its central importance in cell signaling, dysregulation of phosphorylation networks is highly implicated in disease, and kinases represent an important class of drug targets. Mass spectrometry (MS)-based proteomics has enabled significant advances in defining the human phosphoproteome with ever-increasing depth. However, a complete understanding of how phosphorylation orchestrates biological function requires spatial and temporal resolution that remains technically and analytically challenging to obtain. To enable spatial and temporal resolution of the phosphoproteome, we are developing enzymatic tools for covalent capture of phosphorylation marks.
Deciphering the biological functions of unusual post-translational modifications
The human proteome contains numerous rare but evolutionarily conserved post-translational modifications that are only known to occur on a handful of proteins. We are using a multi-pronged approach focused on the development of chemical and enzymatic probes of these modifications to catalog their targets and to elucidate their biological functions.